
Title: Demographics of physics education research
Authors: Stephen Kanim & Ximena C. Cid
First author’s institution: New Mexico State University
Journal: Physical Review Physics Education Research, 16, 020106 (2020)
Picture an introductory physics classroom. What do you think of?
Perhaps you envision a large lecture hall with a professor speaking to hundreds of students, many of whom are white or male. Maybe it’s a small active learning classroom with multiple learning assistants and teaching assistants moving around the room.
While these are possible descriptions of a university introductory physics classroom, most students take introductory physics in high school. If you teach high school physics, maybe you already knew this and pictured a high school classroom from the start.
As you might have assumed, what we are familiar with influences our first guesses. It’s not too hard to stretch that to research: who we are familiar with is who we study. That’s the key result of today’s study, which finds that PER subjects are overwhelmingly not representative of who takes physics. Most subjects come from large, selective, research institutions or do not match the overall demographics of college students.
To reach their conclusion, the authors analyzed PER papers across 45 years from three journals (the American Journal of Physics, The Physics Teacher, and Physical Review Physics Education Research). For each study, the authors documented the type of research study, the total number of students included, the institution the study was conducted at, and the courses the students were enrolled in. If the study did not report these or if the study didn’t focus on students (e.g. a study of textbooks or a theoretical study), the authors excluded it from their sample.
Of the 1,031 papers published in these journals during the time period of interest, only 417 met the criteria established by the authors, which represented over a quarter of a million students.
From these 417 papers, the authors reached six major conclusions about who we study in PER.
1. High school physics is mostly excluded from PER
Even though 3 out of 4 U.S. students studying introductory physics are doing so in high school, only 1 in 10 research subjects are. If one 1971 study is excluded, that drops to 1 in 20.
While the authors note that the specifics depend on which journals they included, the overall result is unchanged. PER tends to ignore high school physics despite the large number of students enrolled in such courses.
2. Physics students from two-year colleges are also largely excluded
Of the college students who take physics, nearly 1 in 4 do so at a two-year college. Yet, only 6 of the over 400 papers in this study included two-year college students. That works out to around 700 students or less than the typical enrollment of an intro physics course at a major research university across 45 years of research.
3. When we do study introductory physics, we tend to focus on calculus-based physics
Only 1 in 3 students who take introductory physics take calculus-based introductory physics. Yet, 4 out of 5 PER subjects were enrolled in calculus-based physics (fig 1).
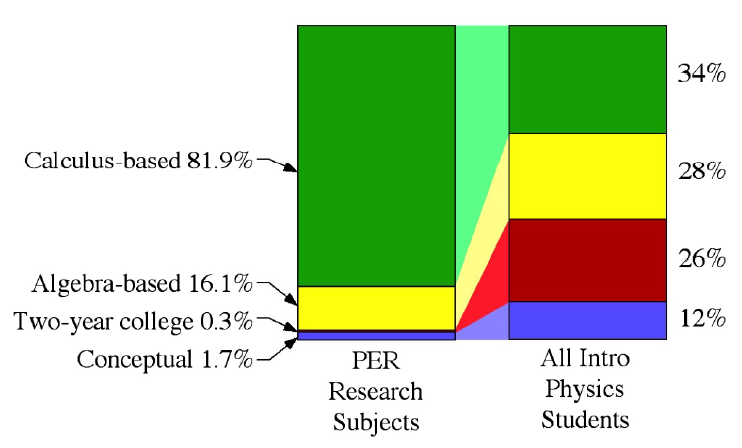
This discrepancy suggests that we are studying students who are more mathematically prepared than typical students in introductory physics. Further, physical science and engineering majors typically take calc-based physics while life science majors tend to take algebra-based physics. Thus, there may be further differences beyond math preparation in who we study and who we teach.
4. PER tends to focus on more mathematically prepared students in general
For this part of their analysis, the authors looked at the SAT math scores of the institutions included in the study. While SAT scores are not the best measures, they are standard for this type of analysis so it’s what the authors used.
Nearly all of the research subjects came from 39 universities so the authors focused on these universities.
The authors then compared the 25th and 75th SAT math score percentiles for these 39 universities to the overall score distribution (fig 2).
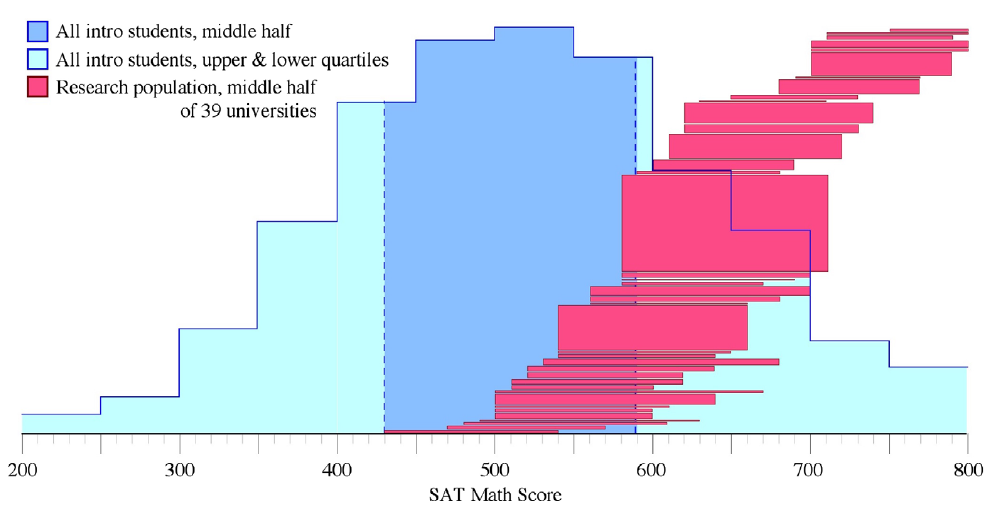
They found that the universities included in PER studies tend to have much higher SAT math scores than the overall college population. In fact, the 25th percentile of these universities corresponds to the 75th percentile nationally.
While not all of the students at these universities will take physics, these results suggest that the students we study in PER are not representative of the larger college population when it comes to math preparation.
5. PER subjects are whiter and wealthier than the typical physics student
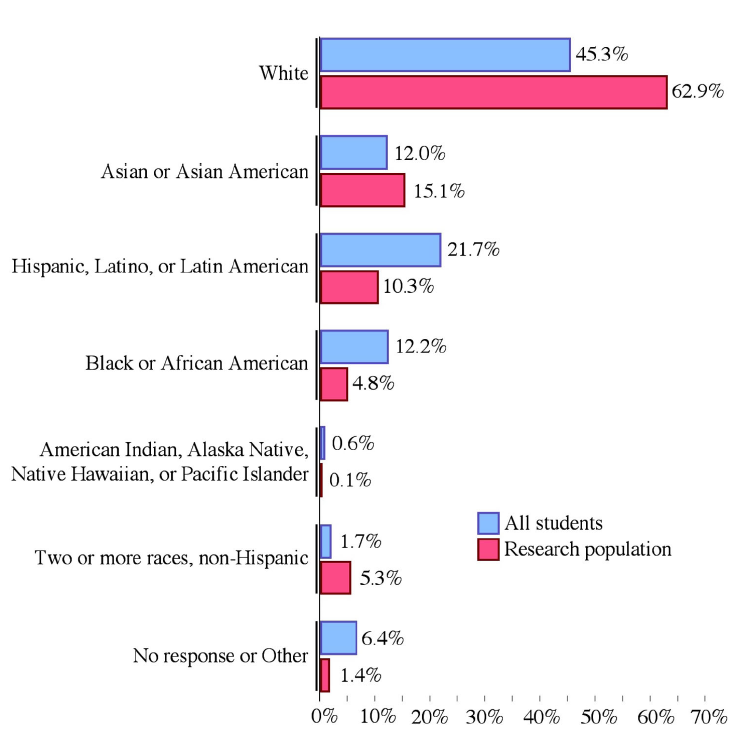
PER studies tend to use convenience samples and since many PER researchers are at primarily white institutions, many of the research subjects are white.
To estimate the racial identities of the subjects in the studies, the authors assumed the racial makeup of physics courses was similar to the racial makeup of the university overall.
The authors found that there are many more white students in PER studies than in the college population overall (fig 3). In addition, while 1 in 3 college students identify as Black, Latinx, or Indigenous, only 1 in 6 PER subjects do.
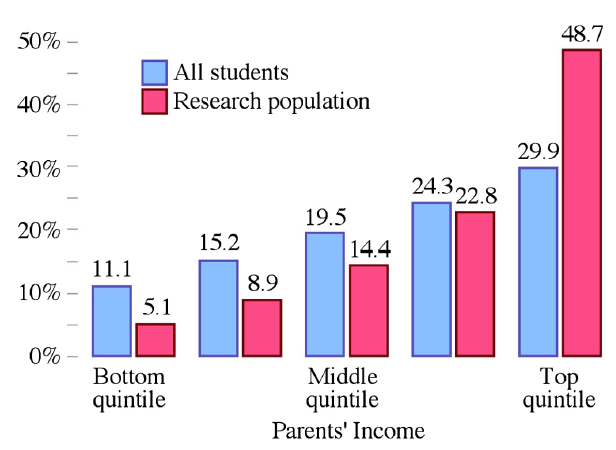
Likewise, the authors then used the income distribution of each university to estimate the income distribution of PER subjects.
Nearly half of the research subjects came from the top 20% of households by income while only 20% of the research subjects came from a household in the bottom 60% of income earners (fig 4).
6. Upper division courses aren’t any better when it comes to representation
Of the research subjects in upper-division physics courses, 4 out of 5 came from only 2 universities and nearly all came from one of 9 universities.
These students again tended to have stronger math preparation and are whiter and wealthier than the general college population. In addition, because studies of upper-division students occur at only a few universities, how representative these studies are is an open question.
What can we do?
The results of this study suggest that the students who we include in our PER studies are not representative of the students who take physics. This doesn’t mean that the results are not useful, but that we should apply caution when applying them to students different from those we study.
Going forward, the authors propose a few recommendations to make sure our studies are broadly applicable or acknowledge when they are not.
- When publishing a PER study, describe the demographics of the students in the study. Including who was studied provides context for the results.
- When starting a study, think about who will be included in the study subjects. Does the study use a diverse sample or does the study only focus on a subset of those who study physics?
- Do more replication studies. While this is good science generally, we need to ensure that our results generalize beyond the students in the study. That our results generalize to all students is often an unstated assumption of PER but we need to ensure that it is actually true.
- Reframe the discussions around race and privilege in PER. PER studies often use a deficit framing where non-white, non-male students are compared to white, male students and the researchers try to explain why the non-white, non-male students perform differently. Instead, the authors of today’s study recommend acknowledging these differences but don’t try to make these students more like “norm” students of PER.
Figures used under CC BY 4.0. Header image by Pexels from Pixabay.
I am a postdoc in education data science at the University of Michigan and the founder of PERbites. I’m interested in applying data science techniques to analyze educational datasets and improve higher education for all students